《制药设备计算机化系统验证指南》速读(Final)
2023-12-11 46
Q1.这是个什么性质的文件,有强制执行性吗?
Q2.谁发布及起草了这个文件,是行业组织还是官方?
Q3.这个文件的适用范围是什么,相关引用文件有哪些?
Q4. 怎样理解文件中的“制药设备验证”,与中国GMP相关要求如何统一?
Q5. 制药设备计算机化系统验证的“全生命周期”方法论
Q6.立项阶段 - 制药设备计算机化系统验证需要考虑的问题
Q7.项目阶段 - 制药设备的CSV(计算机化系统验证)
本文将重点讨论大家都关心的CSV-计算机化系统验证,以及制药设备计算机化系统验证的运行阶段与退役阶段:
Q7. 实施阶段 - 制药设备计算机化系统验证如何起草方案及执行?
大侠解读:
章节6.3系统实施,同样基本参考了 ISPE GAMP5 System Life Cycle Project Phase【也就是我们常说的GxP系统上线过程的计算机化系统验证】,以下图为例
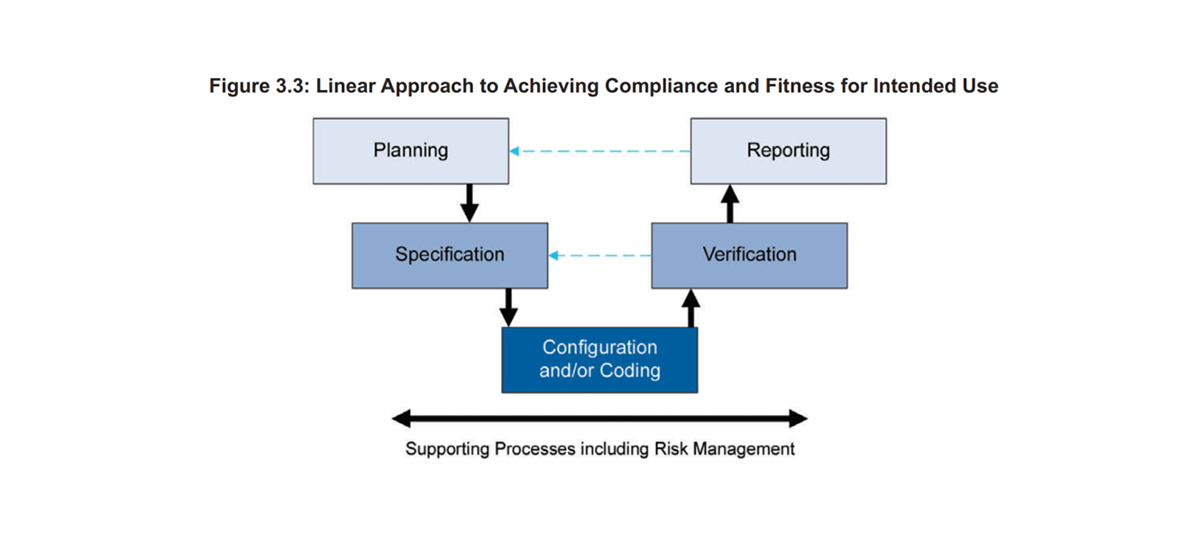
章节6.3.1 计划阶段:
主要工作是评估和定义在整个系统交付及验证过程中的所有交付活动,职责分工以及工作流程。计划阶段的主要交付物为验证计划Validation Plan 和 供应商评估 Vendor Assessment。具体可以参考GAMP5 Appendix M1 – Validation Planning 和 Vendor Activity。

图1 ISPE GAMP5 供应商GxP符合性评估举例
章节6.3.2 设计阶段:
主要工作是确保对于系统的用户需求收集、硬件配置、功能设计、流程配置以及定制化开发过程清晰可追溯,符合系统预期用途。计划阶段的主要GxP验证交付物为规范类文件,如URS、FS、CS和DS等,对于不同复杂程度的系统和在不同GxP质量体系下,对于规范类文件可以有不同要求,比如简单标准化的云系统可以将产品手册作为FS,而某些GxP客户也可能将HDS(Hardware Design Specification)和SDS(Software Design Specification)从CS(Configuration Specification)中独立出来。具体可以参考GAMP5 Appendix D1 & D3 – Specifying Requirement。

图2. 对于自动化制药设备习惯有HDS和SDS,但对于MES制造管理系统则以CS统称
章节6.3.3&6.3.5 测试及确认阶段:
主要工作是确保项目所有要求的规范(Specification)经配置或开发后,经过测试(Testing),已经实现并符合要求(Fulfill GxP Intended Use)。由于软件系统的测试及确认一旦来说同时也是项目验收的重要节点,因此在GxP系统上云软件测试阶段基于不同目的(商务合约付款节点 or GxP CSV验证交付物),进行不同类型的软件测试(非GxP CSV软件测试 vs GxP CSV验证交付物)。具体可以参考(附录)D5 Testing of Computerised System。
| 测试名称 | 是否用于GxP | 备注 |
|---|---|---|
| Unit/Module/Integration/system test单元、模块、集成、系统测试 | 一般Non-GxP,一般使用软件公司标准模板 | IT软件行业最佳实践,属于软件开发阶段,不大适合 GxP CSV |
| FAT/SAT(Factory/Site Acceptance Test)工厂验收测试/现场验收测试 | 一般Non-GxP,可以使用自动化公司标准模板或客户定制化模板 | 设备行业最佳实践,适用于ISPE C&Q 调试与确认工程方法论,与CSV验证方法论有一定差异 |
| S(A)T/UAT (System/User Acceptance Test))系统验收测试/用户验收测试 | GxP CSV可以引用或者替代,但需要使用客户GxP模板 | IT软件行业最佳实践,属于项目交付阶段,与GxP CSV阶段类似 |
| IFQ (Infrastructure Qualification) IT基础设施确认 | GxP CSV常见验证交付物之一 | 网络、存储、安全、平台的IT基础设施确认 |
| IQ/OQ/PQ(installation/operation performance qualification)安装、运行、性能确认 | GxP CSV常见验证交付物之一 | IQ – 客户端服务器硬件是否已经配置到现场,软件是否安装好;OQ – 软件功能是否可以运行,类似上文S(A)T,由项目组在验证环境进行;PQ – 系统是否满足实际业务需求,类似上文UAT,由真实用户在生产环境进行 |
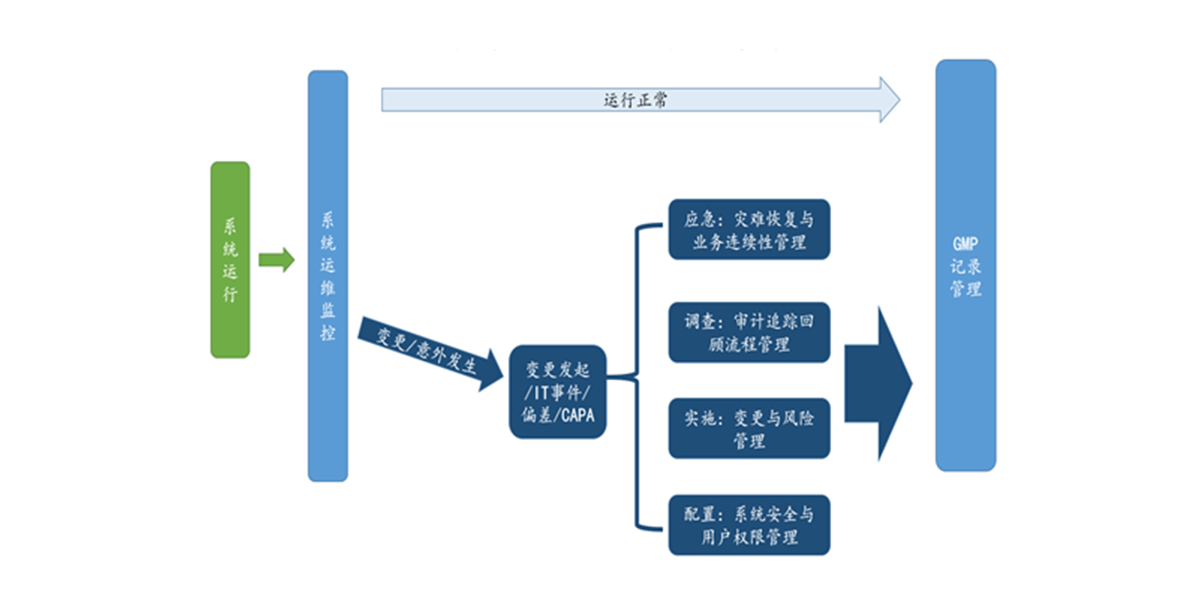
Q8. 运行阶段 - 制药设备的计算机化系统管理注意事项:

大侠解读:
对于制药设备的计算机化系统管理的运行阶段而言,其管理核心主要有以下两点:
- 如何确保计算机化系统上线使用后,始终维持在其当初验证受控状态;包括了人员持续培训、系统管理手册,设备维护日志以及对于计算机化系统要求的周期性回顾(而非直接再验证)。
- 如果检验受控状态被破坏,无论是人为主动(变更)、还是意外异常(偏差),怎么评估风险与恢复正常?怎么确认变更或者偏差未关闭期间系统正常运行、数据符合要求?如何保证变更或者偏差的正常关闭?
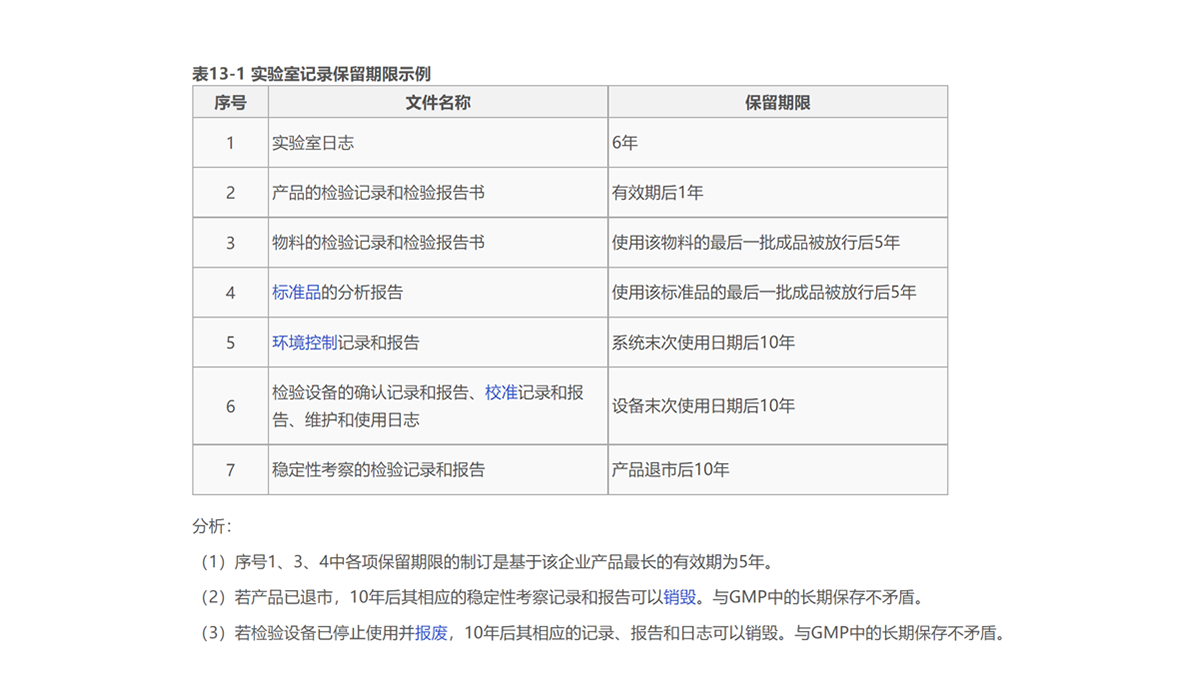
Q9. 退役阶段 - 制药设备如何按要求退役,保证其内所存数据不受影响?
大侠解读:
计算机化系统退役阶段的合规要求主要关注当某个设备、仪器或者IT系统不再被正常维护而退出使用后,其在保存期限内的GxP数据或记录有无按要求正确的迁移到新系统或者归档到安全位置以供监管方未来可能的检查。
GxP记录保留期限和保留要求
以下图为例,为某GMP药企内不同性质的记录或者数据的保存期限;如果某个存有相关记录或者数据的设备、仪器或系统在这个期限内退出现役,其记录或者数据的可读性和安全性必需不受影响。
同时系统退役后,其数据保存应符合数据可靠性要求,在数据保存期限内,可以方便的检索,查看,不可随便修改;因此可以考虑数据归档进入服务器,应用软件虚拟化的方式,以一个便捷和安全的方式满足监管未来的查阅需求。
进一步区分GLP、GCP和GMP不同业务阶段
(参考链接),其数据保存期限从左到右:
